Fragment Network REST API

Tim Dudgeon 2020-08-31
POST
In my last post I described the basic details of how the fragment network is composed and how it allows queries for similar molecules that are “chemically intuitive”. Here I show how queries can be executed through the REST API and how this has turned out to be extremely useful in the virtual screening work we are doing on the SARS-Cov-2 main protease in collaboration with the Diamond Light Source.
The background to this is the X-ray fragment screening program that was initiated at the Diamond Light Source in the early stages of the COVID-19 pandemic and has since progressed at breathtaking speed. Details can be found here.
The main protease is a cysteine protease and a number of covalent and non-covalent fragments were rapidly identified. This work concerns the non-covalent fragments of which there are currently 23 that bind in the active site region. An example is Mpro-x0874_0:
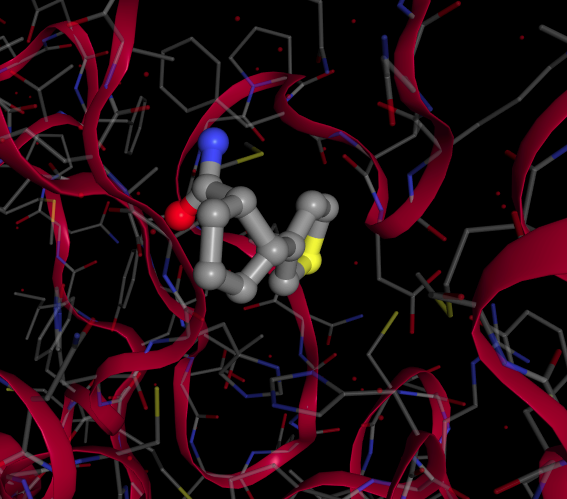
Each fragment is an insight on how a molecule can bind to the protein and is a potential starting point to generating drug leads. To facilitate this we wanted to identify a large number of related molecules that kept the key binding features but expanded the fragment so that it made additional interactions with the protein, and hence might bind more strongly.
Expanding each fragment hit using the fragment network is key to this. Each fragment is a relatively small molecule and we want to expand it but at the same time retain the key features that allowed it to bind to the protein.
As described in the first post of this series this could in principle be done by a skilled chemist using a series of substructure searches, but this would be technically difficult and time consuming for just one molecule, let alone 23 of them.
Instead we use the REST API of our Fragnet Search application. Each fragment molecule can be used as a query and the response are the molecules in the fragment network that are related to the query according to the parameters used. There are 3 key parameters:
- Number of hops - the number of graph traversals to allow from the query molecule (HOPS)
- Change in heavy atom count (HAC)
- Change in ring atom count (RAC)
As we want to find molecules that are bigger than the original fragments we tune the HAC and RAC setting to allow the resulting molecules to be bigger than the query. A value of 1 for HOPS only allows simple additions or deletions so would not give us very interesting molecules. A value of 2 allows more complex changes, including ring and linker substitutions and is typically a good choice. A value of 3 allows more extreme changes still.
The REST API is described here.
This is how you would run an expansion for one of the fragment molecules. The query can be defined in SMILES or Molfile formats. The molecule is standardized on the server before a cypher query is executed against the Neo4j database.
First we need to authenticate against our Keycloak SSO server (using the OpenID Connect protocol).
$ export token=$(curl -d "grant_type=password" -d "client_id=fragnet-search" \
-d "username=username" -d "password=password" \
https://squonk.it/auth/realms/squonk/protocol/openid-connect/token 2> /dev/null | jq -r '.access_token')
Username and password would of course be replaced with real values!
This generates an access token which is stored in the token variable. This token is then provided when we run the query:
$ curl -LH "Authorization: bearer $token" \
"https://fragnet.server/fragnet-search/rest/v2/search/expand/NC(%3DO)%5BC%40H%5D1CCC%5BC%40H%5D1C1%3DCSC%3DC1?hacMin=0&hacMax=8&racMin=0&racMax=6&hops=2"
You would replace fragnet.server with the real server address. The part of the URL between the last / and the ? is the SMILES of the query molecule URL encoded (in this case the actual SMILES was NC(=O)[C@H]1CCC[C@H]1C1=CSC=C1)
That particular query finds molecules that and 1 or 2 graph traversals from the query, have up to 8 additional heavy atoms and up to 6 additional ring atoms.
The response is in JSON format and a small snippet looks like this:
{
"query": "MATCH p=(m:F2)-[:FRAG*1..2]-(e:Mol)<-[:NonIso*0..1]-(c:Mol)\nWHERE m.smiles=$smiles AND e.smiles <> $smiles AND m.hac - e.hac <= $hacMin AND e.hac - m.hac <=
$hacMax AND m.chac - e.chac <= $racMin AND e.chac - m.chac <= $racMax\nRETURN p LIMIT $limit",
"parameters": {
"racMin": 0,
"smiles": "NC(=O)C1CCCC1c1ccsc1",
"hacMin": 0,
"hacMax": 8,
"limit": 5000,
"racMax": 6
},
"refmol": "NC(=O)C1CCCC1c1ccsc1",
"resultAvailableAfter": 5,
"processingTime": 61,
"pathCount": 655,
"size": 633,
"members": [ ... ]
}
That shows metadata about the query. The contents of the members array is elided for brevity. That contains each of the result molecules, an example of which looks like this:
{
"smiles": "C=C1CCC(NC(=O)C2CCCC2)CC1",
"props": {
"inchik": "JMLDPLQMUGOPHE-YHMJCDSINA-N",
"chac": 11,
"hac": 15,
"inchis": "InChI=1/C13H21NO/c1-10-6-8-12(9-7-10)14-13(15)11-4-2-3-5-11/h11-12H,1-9H2,(H,14,15)/f/h14HMA"
},
"cmpd_ids": [
"MOLPORT:044-550-146"
]
}
That was really just for illustrative purposes. In the real world we wrap this up in a simple Python script that can be found here. This reads a SD-file containing all the fragment molecules, runs a query for each one, and provide outputs in a suitable format for the first step of the virtual screening process.
Running that script typically takes only a few seconds, and you usually get a few hundred or a few thousand candidates for each query in a format ready to screen. The number depends on the query molecule and the query parameters.
All those candidates are related to the query in a chemically intuitive way. The expansion is fast and doesn’t need a skilled chemist.
If you are interested in using the Fragment network then get in touch with us.
latest posts
by year
2024
2023
2021
2020
Fragment Network REST API Cookie-cutting Ansible Kubernetes Projects Deploying container images from a private GitLab registry Fragment network basics Fragment network intro Redirecting to www with an nginx ingress Installing Kubernetes with Pharos Virtual screening for SARS-Cov-2 main protease inhibitors
2019
2018
by category
Blog
Containers
Software design
Automation
AWS ParallelCluster v3 Custom Images Migrating to AWS ParallelCluster v3 Kubernetes object linting with popeye GitHub Actions for container images Cookie-cutting Ansible Kubernetes Projects Installing Kubernetes with Pharos Building machine images with Packer Python and the Jenkins API Applying the build process to the deployment
IaC
Docking
Fragment network
Kubernetes
Kubernetes PreStop Lifecycle Hooks A kubernetes volume replicator Fixing a broken etcd cluster Kubernetes object linting with popeye Cookie-cutting Ansible Kubernetes Projects Deploying container images from a private GitLab registry Redirecting to www with an nginx ingress Installing Kubernetes with Pharos
Web
Ansible
Python
Squonk