Fragment Network
More up to date information is available on our Fragnet Search site. This page is retained for historical purposes.
About the Fragment Network
The fragment network was conceived by Astex as an approach to identifying follow up compounds from fragment screening. It is described in this paper published in 2017.
The network is built from a set of molecules by recursively fragmentation based on their ring systems and the substituents of those rings. That network forms a directed acyclic graph that can be traversed to identify similar molecules based on sharing common fragments. Compared to traditional chemical fingerprint similarity approaches the fragment network has a number benefits:
- More intuitive to chemists
- Similarity is more meaningful
- Better for small molecules where few fingerprint bits will be set
Implementation
The Astex paper described the methodology nicely, but the code was not released and was based on the Daylight toolkit. Anthony Bradley at Diamond saw the potential of the methodology for fragment screening and re-implemented it using the RDKit chemical toolkit. His implementation can be found here. Python modules are used to build the fragment network data (the nodes and edges of the graph) which is then loaded into a Neo4j graph database that can be searched. That database is then used in their Fragalysis application which Informatics Matters is helping deploy and manage.
Informatics Matters are working with Diamond to extend the methodology and make it accessible to other parties and for use outside of fragment screening.
Datasets
Our methodology allow to build fragment network data for a number of input datasets and then to combine these into a searchable Neo4j database. Private datasets can be processed for customers on demand. Example datasets that we have built to date include:
| Source | # compounds | # nodes | # edges |
|---|---|---|---|
| Molport | 7,486,593 | 107,899,273 | 607,806,848 |
| Enamine REAL DSI poised library rule of 4 | 1,394,963 | 5,380,055 | 24,439,600 |
| Enamine REAL DSI poised library rule of 5 | 39,765,321 | 191,230,680 | 1,038,917,131 |
| SENP7 HTS dataset | 330,688 | 6,949,173 | 36,188,234 |
| ChemSpace building blocks | 5,535,472 | 10,124,955 | 40,526,531 |
Building these datasets requires a lot of computational power, but once built the database can be searched very efficiently.
Search interfaces
We have a number of ways of searching the fragment network database:
Fragnet Search
Fragnet Search is our new web interface for searching the fragment network. This allows intuitive searching the fragment network, analysing the results based on the type of transform along with collecting lists of compounds to purchase from the provided vendors.
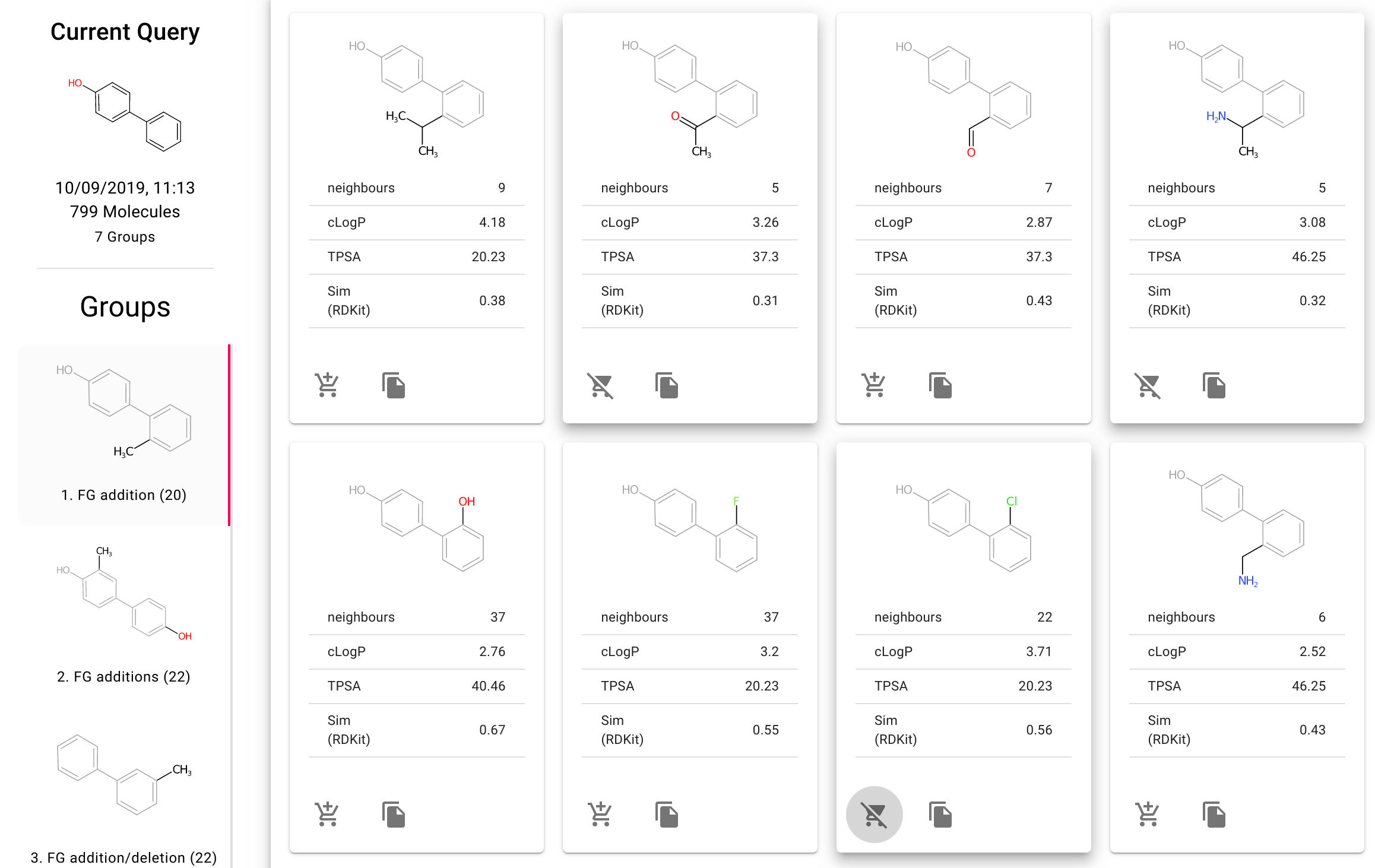
REST API
This allows to run queries against the database (e.g. using a query molecule in SMILES format) and get back the set of similar molecules grouped by the type of transformation (e.g. all molecules with a substitution at each position). A number of calculated molecular properties can be included in the results.
Optibrium's Stardrop
We have integrated the REST API into Optibruim's Stardrop desktop chemistry application. Using a simple query UI you can search the fragment network and display the results in Stardrop's CardView, with molecules of the same transformation being grouped in the same stack. This can be seen in the following screenshot:
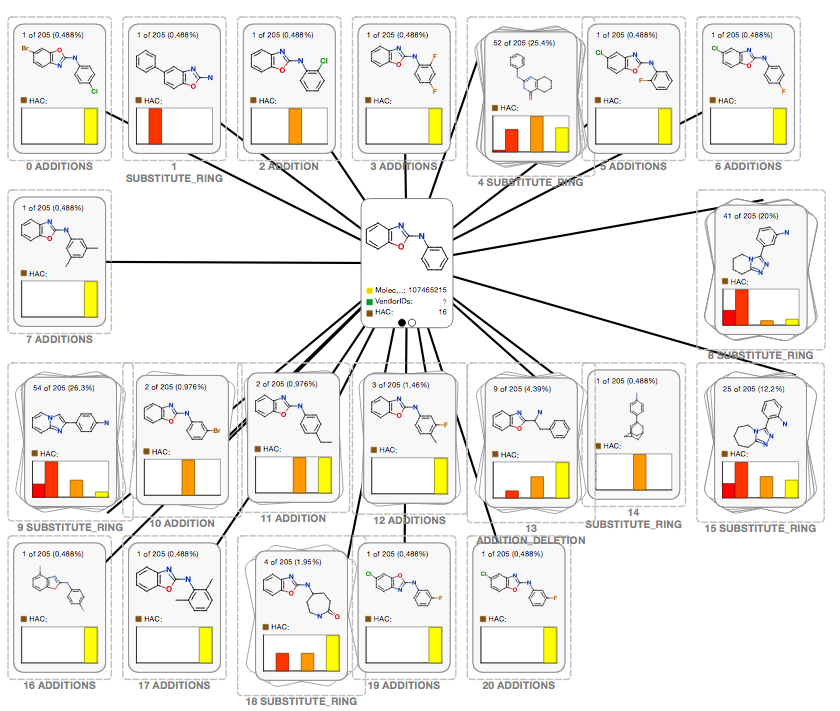
Access and availability
Access to the hosted REST API running on Amazon Web Services is provided on a subscription basis.
The Stardrop integration is provided through Python scripts that are freely available, but require a subscription to the hosted REST API.
Building custom databases and setting up the database and search API on your own infrastructure can be provided through our consultancy services.
For more information on any of these please contact info@informaticsmatters.com